Electronic effects in irradiation damage
State of the art simulations of radiation damage incorporating electron-ion energy exchange
Almost all simulations of collision cascades make the Born-Oppenheimer approximation: they assume that the electrons respond so quickly to the moving atoms that they remain always in the ground state. This approximation is implicit in the potentials used in classical molecular dynamics (MD). But in reality the ions in collision cascades move so fast that the electronic system cannot keep up; the electrons become excited and exert additional non-adiabatic forces on the ions. These non-adiabatic forces reduce the amount of energy in the initial cascade and the hot electrons either transport energy away from the cascade (quenching in defects), or act as a heat-bath (annealing out defects).
The electronic frictional force and electronic stopping
We propose a new model for the electronic frictional forces in radiation damage collision cascades in metals, which successfully captures the complex directionality and local-environment dependence revealed in quantum-classical simulations, but takes a time- and space-local form, suitable for inclusion in classical molecular dynamics simulations at very low computational cost. This model makes it possible to include the effects of electron-ion energy exchange within classical simulations of radiation damage with unprecedented accuracy.
In an attempt to capture the effects of electron-ion energy transfer many researchers have augmented the ionic forces in MD simulations with additional electronic friction forces. These most often take the form of a simple drag force, directly opposed to and proportional to each ion's velocity. In fact, our previous simulations of radiation damage, using a time-dependent tight-binding model of a generic metal evolving under quantum-classical Ehrenfest dynamics, have shown that the friction force is richly structured. It depends sensitively on the position, velocity and local atomic environment of each moving ion, is inherently many-atom and is long-ranged and history-dependent. Critically, it is not, in general, well approximated by a simple drag.
Starting with the equations of motion for a system of classical ions coupled to a set of quantum mechanical electrons we have derived a new classical expression for the electronic frictional forces, which captures much of their rich behaviour. Our expression is time- and space-local and has a physical interpretation in terms of a lagged bond forming and breaking response of the moving atoms. The expression takes into account the local electronic structure within the second-moment approximation and makes use only of quantities (instantaneous ionic positions and velocities) easily accessible in existing MD codes. As such, it represents a way to capture much more of the physics of electron-ion energy transfer in large-scale classical simulations than has previously been possible.
In an attempt to capture the effects of electron-ion energy transfer many researchers have augmented the ionic forces in MD simulations with additional electronic friction forces. These most often take the form of a simple drag force, directly opposed to and proportional to each ion's velocity. In fact, our previous simulations of radiation damage, using a time-dependent tight-binding model of a generic metal evolving under quantum-classical Ehrenfest dynamics, have shown that the friction force is richly structured. It depends sensitively on the position, velocity and local atomic environment of each moving ion, is inherently many-atom and is long-ranged and history-dependent. Critically, it is not, in general, well approximated by a simple drag.
Starting with the equations of motion for a system of classical ions coupled to a set of quantum mechanical electrons we have derived a new classical expression for the electronic frictional forces, which captures much of their rich behaviour. Our expression is time- and space-local and has a physical interpretation in terms of a lagged bond forming and breaking response of the moving atoms. The expression takes into account the local electronic structure within the second-moment approximation and makes use only of quantities (instantaneous ionic positions and velocities) easily accessible in existing MD codes. As such, it represents a way to capture much more of the physics of electron-ion energy transfer in large-scale classical simulations than has previously been possible.
|
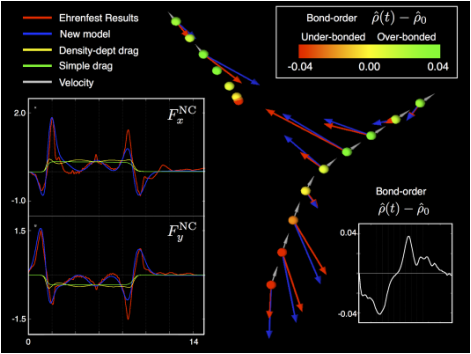
Figure: The main figure shows snapshots (0.25 fs apart) of two atoms undergoing an oblique collision in a quantum-classical Ehrenfest simulation of a 5 keV collision cascade. The red arrows show the electronic friction force from the full simulation and the blue arrows show the predictions of our new model, which reproduce well the complex directionality. The ions are coloured according to the difference between the time-evolved and ground-state bond-order between the colliding atoms. We can clearly see the lag in the bonding response, leaving the atoms under-bonded for the first half of the collision and over-bonded in the second half. The inset figure in the bottom right shows this difference in the bond-order. The inset in the bottom left shows two cartesian components of the force on the target ion. The quantum-classical simulation results are compared to the predictions of our new model, a simple drag force model and a second commonly used model of a local electron density dependent drag. Our new model reproduces much of the complex detail, in contrast to the drag force models.
|
Published in: C P Race, D R Mason, and A P Sutton. "An improved model of interatomic forces for large simulations of metals containing excited electrons." New Journal of Physics, 12, 93049-67 (18pp), 2010.
Electronic excitations, elevated temperature and weakened bonds
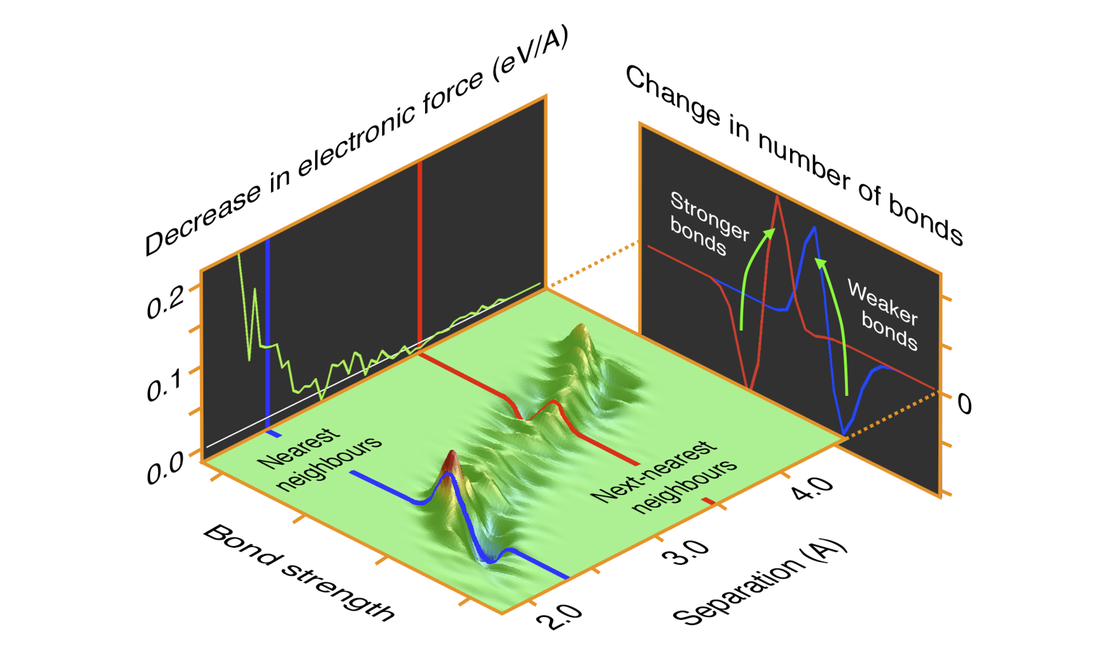
Figure: A double histogram of ion pairs binned according to bond strength and inter-ionic separation shows the bond weakening effect of the excitations for ions with separations close to the nearest neighbour distance. Projections onto each dimension of the histogram highlight these effects.
|
Using time-dependent tight-binding simulations of radiation damage cascades in a model metal we can directly investigate the nature of the excitations of a system of quantum mechanical electrons in response to the motion of a set of classical ions. We we can then calculate the effect of these excitations on the attractive electronic forces between the ions. We find that the electronic excitations are well described by a Fermi-Dirac distribution at some elevated temperature, even in the absence of the direct electron-electron interactions that would be required in order to thermalize a non-equilibrium distribution. We explain this result in terms of the spectrum of characteristic frequencies of the ionic motion. We find that these excitations cause a weakening of the bonding interactions between nearest neighbours (see figure) and that this weakening is mostly (95%) accounted for by a thermal model for the electronic excitations. This result justifies the use of the simplifying assumption of a thermalized electron system in large scale atomistic simulations of radiation damage with an electronic temperature dependence and in the development of temperature dependent interatomic potentials.
|
Published in: C P Race, D R Mason, and A P Sutton. “Electronic excitations and their effect on the interionic forces in simulations of radiation damage in metals.” Journal of Physics: Condensed Matter, 21, 115702 (8pp), 2009.
Resonant charging and stopping power of channelling atoms in a crystalline metal.
We have used quantum-classical Ehrenfest dynamics simulations to investigate the effects of ion-electron energy exchange on the passage of slow heavy ions along open channels in a model crystalline metal. Such ion channelling is exploited in, for example, implantation technologies in semiconductor device fabrication and the hardening of tool steels by nitrogen implantation, but experimental data showing ions becoming charged while passing through foils are not fully understood, perhaps due to the neglect of key physics in models of ion penetration.
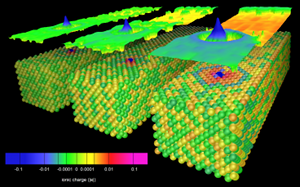
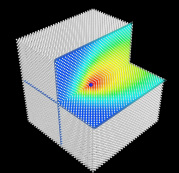
Figure: A channelling heavy ion can become charged when the frequency at which it passes its neighbours matches the difference between Fermi level and its localized states. In this image of the steady state computed for slow ions in a model fcc metal, a self-interstitial ion is moving towards us down a <100> channel with kinetic energy 0 keV (back image), 10 keV (middle) and 365 keV (front). Ions are shaded according to their charge using a power-law colour scale. Above the crystals the Hartree potential in the plane of the ion is also shown. At 365 keV (10.5 A/fs, 0.48 a.u.) there is a resonant maximum in the charge accumulated on the channelling ion.
|
By developing an approach that treats the coupled evolution of a set of classical ions and a system of quantum mechanical electrons, we have been able to directly simulate ion channelling including the effects of ion-electron energy exchange and electronic excitation. By exploiting a simple tight binding model of the electronic structure of a model metal they are able to reach the time and length scales necessary for such simulations and currently inaccessible to more quantitatively accurate methods such as time-dependent density functional theory.
The simulations reveal a resonant accumulation of charge on a channelling ion analogous to the Okorokov effect (a resonant excitation from core to valence states of a single atom) but originating in electronic excitation between delocalized states and localized valence states on the channelling ion and its transient host neighbours. This accumulation is stimulated by the time-periodic potential experienced by the channelling ion and occurs when the frequency at which the ion moves from interstitial point to equivalent interstitial point along the channel matches the frequency corresponding to excitations between the Fermi level and the localized states. The charge resonance reduces the electronic stopping power on the channelling ion. These are surprising and interesting new chemical aspects of channelling, which cannot be predicted within the standard framework of ions travelling through homogeneous electron gases or by considering either ion or target in isolation. |
Published in: D R Mason, C P Race, A P Horsfield, T N Todorov, W M C Foulkes, and A P Sutton. “Resonant charging and stopping power of channelling atoms in a crystalline metal.” New Journal of Physics, 14, 73009 (14pp), 2012.
